
Recherche:Chiralité prébiotique

De l'origine mécanique et géométrique de la chiralité et de l'homochiralité
du glycérol-phosphate, du glycéraldéhyde-P et des acides aminés prébiotiques
Résumé
Le rapprochement des phospholipides, dans la bicouche d'un liposome, provoque leur rotation autour de leur axe d'acides gras, générant une force qui rapproche les 2 feuillets de la bicouche. Dans cette étude théorique je montre que pour obtenir la plus grande cohésion du liposome, par ces forces, la sérine de la tête hydrophile doit avoir une chiralité L. Dans le cas où la tête hydrophile serait absente les acides aminés de chiralité L pourraient contribuer à cette cohésion en prenant la place de la L-sérine. Certains coenzymes présentant une configuration analogue à l'éthanolamine pourraient y contribuer aussi. C'est le cas de la pyridoxamine, de la thiamine et du tétrahydrofolate prébiotiques.
Le regroupement des acides aminés de chiralité L et de la pyridoxamine sur la paroi pourrait initialiser le métabolisme prébiotique de ces seuls acides aminés. C'est ce qui expliquerait l'origine de l'homochiralité des acides aminés dans le vivant.
Par ailleurs je montre que, dans la tête hydrophile, l'estérification du glycérol-phosphate à 2 acides gras passe par le positionnement de la dihydroxyacétone-phosphate et du L-glycéraldéhyde-3-phosphate , mais pas du D-glycéraldéhyde-3-phosphate, avant leur hydrogénation en glycérol-3-phosphate. L'accumulation du D-glycéraldéhyde-3-phosphate dans le cytoplasme déplacerait les équilibres thermodynamiques vers la synthèse de la D-dATP à partir du D-glycéraldéhyde-3-phosphate, de l'acétaldéhyde et de l'adénine prébiotique, réaction qui ne nécessite pas de coenzyme dans le métabolisme biotique. La D-dATP et la thiamine, plus le métabolisme prébiotique des acides aminés L sur la paroi, initialiseraient les voies des D-pentose-phosphates et des D-nucléotides à partir de la réaction D-glycéraldéhyde-3-phosphate + dihydroxyacétone-phosphate et des bases nucléiques prébiotiques.
L'épuisement du glycéraldéhyde prébiotique (racémique) et le métabolisme biotique naissant, dominé par le D-glycéraldéhyde-3-phosphate, expliqueraient l'origine de l'homochiralité des sucres dans le vivant.
Note du 14.03.2015: Cet article fait partie de la synthèse de mes travaux jusqu'en 2014, synthèse publiée dans Origins of Life and Evolution of Biospheres de mars 2015.
Référence: Prebiotic Petroleum; Mekki-Berrada Ali, Origins of Life and Evolution of Biospheres, 2015, DOI 10.1007/s11084-015-9416-7[1].
Introduction
[modifier | modifier le wikicode]17/11/11 fini le 14/02/12 mise à jour le 15/08/13
Dans ce qui suit j’ai abrégé les termes suivants parce qu’ils sont très utilisés:
acide aminé: aa
acide gras: ag
phospholipide: PLD
phosphate: P.
Cette étude théorique est faite dans le cadre de l'hypothèse d'une évolution moléculaire prébiotique qui se déroulerait dans une poche de pétrole abiotique d'origine géochimique ( pétrole prébiotique ). Le développement théorique et bibliographique de cette hypothèse a montré qu’il était tout à fait possible que les liposomes puissent se former dans ces conditions à partir de vésicules aqueuses présentes dans la phase huile et qui migreraient dans la phase eau pour former ces liposomes. La paroi des vésicules seraient formées par la tête carboxylique des acides gras et le contenu en eau de ces vésicules, ainsi que la phase eau principale, contiendraient les petites molécules hydrophiles dont le glycérol, l'éthanolamine, le phosphate et certains acides aminés racémiques, dont la sérine, pour synthétiser par estérification la tête hydrophile que l’on connaît.
C'est en essayant d'imaginer l'agencement de ces petites molécules en tête hydrophile (dans la vésicule ou le liposome) et la fixation de cette tête sur les ag, qu’il m’est apparu que ce processus puisse imposer la chiralité du glycérol-P et des aa ainsi que la séquestration de ces aa sur les parois, en fonction de leur chiralité, mais aussi de leur longueur, de leur volume et de la fonction chimique portée par leur radical.
Dans un premier temps nous étudierons l’enchaînement des molécules de PLD, les unes aux autres, grâce à l'éthanolamine ou à la choline ou à la sérine, qui font partie intégrante de la tête hydrophile. Nous montrerons que cet agencement par enchaînement est à l'origine de la chiralité de la sérine. Nous étudierons ensuite la chiralité du glycérol-P dont l'origine est à trouver dans un compromis entre l'encombrement stérique et la répulsion électronique entre la tête carboxylique de l'ag proche du P et ce dernier.
Un chapitre particulier sera dédié à la séquestration des aa libres parce qu’ils vont se mettre à la place des têtes hydrophiles, grâce à leur amine, lors de l'agencement dans les vésicules ou lors de la perte de ces têtes en réaction au milieu extérieur.
Nous terminerons enfin par les propriétés de cette homochiralité de la paroi liposomale qui peut être à l'origine de l'initialisation du métabolisme prébiotique dans la bicouche même, hypothèse émise dans le travail sur la chimio-osmose prébiotique. L'homochiralité du métabolisme que nous connaissons serait originaire de celle de la bicouche prébiotique.
Chiralité de la serine prébiotique
[modifier | modifier le wikicode] La serine est attachée par liaison ester au phosphate comme le sont l'éthanolamine, la choline et le glycérol. Cette partie de la tête hydrophile constitue un bras mobile. La 2e partie de la tête hydrophile est le bras fixe, constitué du glycérol-P fixé à 2 ag. Même quand la molécule de PLD est isolée des autres molécules de PLD, le bras fixe présente une grande inertie. Et lorsque 2 molécules de PLD se rapprochent par la force de la liaison hydrogène (NH2) ou ionique (NH3+), vers les 2 oxygènes libres du P, et par la force de l'hydrophobicité (rapprochement des 4 ag), c’est le bras libre qui bougera en premier. Et ceci d'autant plus vite et plus fort que la liaison hydrogène (ou ionique) agit à plus grande distance que ne le font les forces de Van der Walls pour l'hydrophobicité des ag. Ceci entraîne obligatoirement une rotation de la molécule de PLD entière, autour des 2 ag, car l'amine NH2 (NH3+) n’est pas alignée avec le bras libre, et le P dont elle se rapproche est massif et très proche des ag.
Si l’on se positionne au-dessus du PLD avec les ag en dessous du plan défini par 3 oxygènes du P, une rotation anti-horaire soulèverait la molécule de PLD et une rotation horaire l'enfoncerait. La rotation horaire rapproche donc le feuillet en question vers l'autre feuillet de la bicouche, ou vers la phase huile pour la vésicule aqueuse. La rotation horaire est donc un facteur de cohésion très important si l’on tient compte du grand nombre de molécules de PLD que contient la bicouche. Et c’est cet agencement qui déterminera la chiralité de la serine et de la position de l'amine NH2 (NH3+) de l'éthanolamine.
Positionnement du NH2 de l'éthanolamine dans la molécule de phospholipide pour obtenir la plus grande cohésion possible du liposome
[modifier | modifier le wikicode] Si on regarde de face les 2 oxygènes libres du phosphate (voir dessin 1), NH2 peut occuper 4 positions possibles: avant-gauche(VG), arrière-gauche(RG), avant-droite(VD), arrière-droite(RD). La position RG est la seule à donner un enchaînement, avec rotation horaire, d'un grand nombre de PLD.
En effet, les 2 positions VG et RD tournent dans le sens anti-horaire pour rejoindre le P. Les 2 autres positions tournent dans le sens horaire, mais la position VD ne peut s'agencer qu'avec 1 seul autre PLD, car NH2 et les 2 oxygènes du P sont du même côté et se neutralisent mutuellement. Par contre la position RG permet d'agencer à la queue leu-leu et de façon illimitée les PLD les uns aux autres tout en assurant le rapprochement des 2 feuillets du liposome par rotation horaire.
-
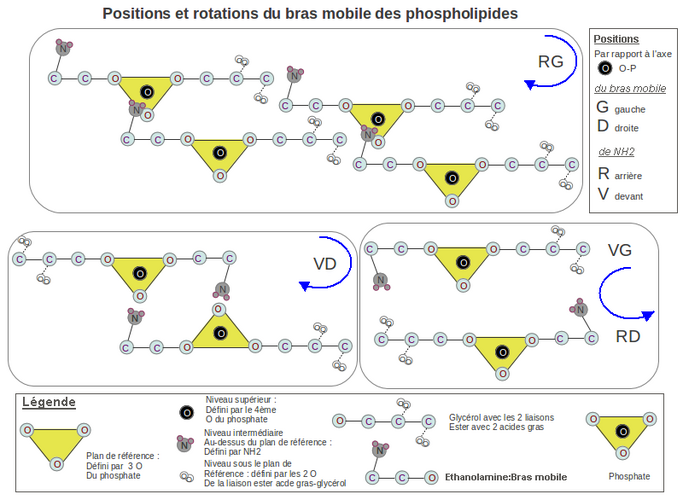 Dessin 1. Hypothèse de l'origine mécanique de la chiralité dans les phospholipides. Ici est traité le positionnement de l’éthanolamine par rapport au phosphate où son déplacement vers lui pour établir une liaison hydrogène entraîne une rotation. Cette hypothèse stipule que la rotation horaire et le positionnement à la queue leu-leu des molécules phospholipides (encadré RG) entraîne le rapprochement des 2 feuillets du liposome, augmentant sa cohésion. Dans cette configuration la sérine aurait une configuration L de type lévogyre, car son carboxyle ne peut se mettre sous le plan de référence défini par le phosphate (voir la légende dans le dessin) et serait alors à proximité des carboxyles des 2 acides gras des molécules voisines.
Dessin 1. Hypothèse de l'origine mécanique de la chiralité dans les phospholipides. Ici est traité le positionnement de l’éthanolamine par rapport au phosphate où son déplacement vers lui pour établir une liaison hydrogène entraîne une rotation. Cette hypothèse stipule que la rotation horaire et le positionnement à la queue leu-leu des molécules phospholipides (encadré RG) entraîne le rapprochement des 2 feuillets du liposome, augmentant sa cohésion. Dans cette configuration la sérine aurait une configuration L de type lévogyre, car son carboxyle ne peut se mettre sous le plan de référence défini par le phosphate (voir la légende dans le dessin) et serait alors à proximité des carboxyles des 2 acides gras des molécules voisines.

La chiralité de la sérine est de type lévogyre (L).
[modifier | modifier le wikicode] Nous venons de voir l'origine mécanique du positionnement du NH2 de l'éthanolamine. Ceci est valable aussi pour le NH2 de la sérine. Le carbone de la fonction carboxylique de la sérine va se positionner, lui, par répulsion électronique. En effet si on dessine l'agencement de plusieurs PLD voisins (en vue aérienne, voir dessin 1-RG), on voit que les PLD d'une rangée s’emboîtent dans la rangée voisine et que le carbone de l'amine (NH2) se trouve au même niveau et aligné le long d'une oblique, avec les carbones portant les esters des glycérols.
Si on positionne la fonction carboxylique de la sérine sous le plan défini par le P (ce qui donnerait la D-sérine), alors elle se trouvera entourée des 4 oxygènes des 2 liaisons ester appartenant aux 2 molécules de PLD successives. Il y aura alors, dans un espace exigu, 6 oxygènes pour un seul hydrogène pour faire la liaison, dans le meilleurs des cas suivant le pH. Comme les 2 fonctions carboxyliques des 2 ag sont fixes, suite à la cohésion globale du liposome, celle de la sérine, mobile, est expulsé automatiquement par répulsion électronique au-dessus du plan défini par le P. Nous avons alors de la L-sérine.
Dans le cas des archées ce n'est plus la répulsion électronique qui intervient, mais l'encombrement stérique. Le carbone de la fonction carboxylique de la D-sérine s'intercalerait entre les têtes des ag, ce qui écarte les 2 PLD de la rangée voisine, augmentant fortement l'encombrement. Or l'encombrement stérique doit être minimal pour ces procaryotes, comme on le verra aussi pour la chiralité de leur glycérol-P du bras fixe.
Chiralité du glycérol-P prébiotique
[modifier | modifier le wikicode] La chiralité du glycérol-P doit être indépendante de la position du bras mobile comme c’est le cas entre bactéries et archées. Ces lignées assurent la cohésion du liposome avec le même principe mécanique qu'on vient de voir, mais les chiralités de leur glycérol-P du bras fixe sont opposées. La chiralité du bras fixe dépend de la liaison du glycérol aux chaines aliphatiques. En effet la liaison ester est volumineuse et répulse les électrons avec ses 2 oxygènes, alors que la liaison éther n'a qu'un oxygène et est moins répulsive que chacun des 2 oxygènes de la liaison ester (la liaison éther est connue pour être très stable, étant très peu réactive).
Chez les archées, avec la liaison éther sur les ag pour minimiser l'encombrement stérique, les 2 liaisons éther peuvent se mettre toutes les 2 du même côté que les 2 oxygènes libres du P, pour remplir l'espace vacant sous le demi-plan de référence défini par le P (voir Dessin 2). Comme la position de la sérine par rapport au P est imposée mécaniquement, comme on l'a vue précédemment, son amine (NH2) et l'oxygène de la première liaison éther se trouvent, du coup, chacun dans un demi-plan différent, la chiralité du glycérol-P est alors de type lévogyre comme la sérine, puisqu’ils se superposent par rotation. C'est un L-glycérol-3P si l'alcool terminal se trouvait au-dessus des carbones du glycérol comme le CO2H de la sérine se trouve au-dessus de la membrane. Or pour diminuer l'encombrement stérique l'alcool terminal du glycérol doit être au-dessous de ses carbones. C'est alors un D-glycérol-3P ou L-glycérol-1P (sn-glycérol-1P), celui qu'on trouve chez les archées.
Avec la liaison ester, la répulsion électronique entre elle et les oxygènes libres du P, positionne automatiquement la première chaîne aliphatique la plus proche du P, du même côté que l'amine de la sérine, de l'autre côté du demi-plan défini par le P (voir Dessin 1-RG). Et par encombrement et répulsion avec la première, la deuxième chaîne aliphatique se trouve du côté du demi-plan contenant le P et sous les carbones du glycérol pour les mêmes raisons d'encombrement que pour les archées. D'où le L-glycérol-3P (sn-glycérol-3P) des bactéries ou D-glycérol-1P (voir KEGG[2] pour les synonymes ).
Il faut aussi signaler que dans la vésicule ou sur le feuillet interne du liposome, lors de la mise en place des têtes hydrophiles, les ag sont collés côte à côte et, l'hydroxyle de l'un faisant face à l'oxygène de l'autre, la liaison hydrogène entre eux se trouve renforcée. Lors de la fixation des têtes hydrophiles sur un champs de carbones carboxyliques, la moitié du travail est faite et reste juste à positionner la tête.
-
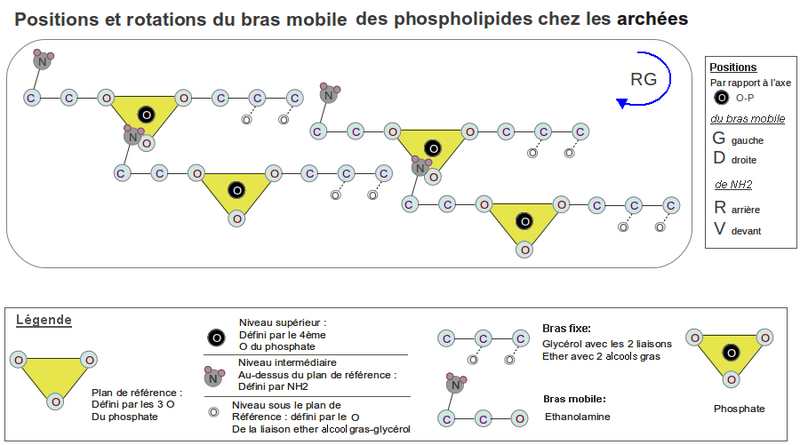 Dessin 2. Voir Dessin 1 pour la légende.
Dessin 2. Voir Dessin 1 pour la légende.

Les caractéristiques physico-chimiques des acide aminés prébiotiques
[modifier | modifier le wikicode]Les acides aminés dans la théorie de la chimio-osmose prébiotique
[modifier | modifier le wikicode] J’ai étudié déjà l’interaction aa-liposome dans l’article théorique sur la chimio-osmose prébiotique. J’ai supposé alors que pour former les pores prébiotiques, qui, à l'instar des ionophores produits par certains procaryotes, contiennent des acides aminés (D et L) et des acides alpha-hydroxylés (D et L), les aa devaient s'intercaler entre le P et l'amine, avec liaison hydrogène des 2 côtés. Leur radical se positionnant du coup verticalement à la bicouche, soit dans la bicouche, soit à l'extérieur.
Le passage des aa à travers la bicouche, par des pores prébiotiques ou unitairement, m'a conduit à supposer que le métabolisme pourrait bien commencer dans la bicouche qui leur sert alors d’échafaudage pour les positionner les uns à côté des autres. L'efficacité de ces groupements d'aa serait de plus en plus grande si leur homochiralité augmente de plus en plus. Mais je n’avais émis aucune hypothèse sur le type, D ou L, de la chiralité finale de ces aa.
De même, la ressemblance de 13 aa biotiques avec l'éthanolamine et la sérine pour la longueur de leur chaîne carbonée (2 C) entre la tête et la fonction portée par le radical, ainsi que les contraintes qu’ils doivent subir lors de leur passage à travers la membrane, me laissaient deviner que les aa prébiotiques devaient être courts et hydrophobes. Par contre les radicaux ionisés ou très réactifs posaient problème pour le passage à travers la membrane. Ce sont les radicaux ionisables: acides, amides, amines ainsi que ceux de l'histidine et de l'arginine.
La séquestration des aa dans la théorie de la cohésion mécanique du liposome, que nous allons voir juste après explique leur origine chirale L, origine que n'arrive pas à introduire la théorie de la chimio-osmose prébiotique. Cependant les 2 processus, diffusion à travers la membrane et séquestration des aa que nous allons voir tout de suite, ne sont pas exclusifs l'un l'autre et peuvent coexister en même temps.
La séquestration des acides aminés dans la théorie de la cohésion mécanique du liposome
[modifier | modifier le wikicode]Principe:
[modifier | modifier le wikicode] Nous avons vu précédemment que la chiralité de la sérine dans le liposome a son origine dans une force qui rapproche les 2 feuillets. C'est une force perpendiculaire à la membrane. Mais elle n'est possible que si la sérine est rattachée au P du bras fixe par une liaison ester. D'ailleurs toute amine rattachée au P peut servir cette cohésion. C'est le cas de l'éthanolamine, de la choline mais aussi du glycérol avec son hydroxyl terminal pour la liaison hydrogène.
Cependant la cohésion mécanique du liposome a 2 composantes, la composante perpendiculaire à la surface du liposome que nous venons de voir et que je nommerai verticale, mais surtout une 2e composante perpendiculaire au plan défini par les 2 ag du PLD et que je nommerai horizontale, plus forte, et dont le sens va de l'amine d'un PLD au P du PLD suivant. Il n'y a pas de force de cohésion perpendiculaire à la force horizontale et contenue dans le plan des ag d'un même PLD. C'est ce qui autorise les glissements des chaînes de PLD les unes par rapport aux autres et différencie le liposome de la rigidité du cristal.
La cohésion horizontale a 2 composantes parallèles, la première, la liaison hydrogène, amine-P, entre 2 têtes hydrophiles consécutives et la deuxième, constituée par les liaisons de Van der Walls entre les queues aliphatiques des 2 PLD consécutifs. Si ces dernières constituent le fondement même de la formation des vésicules, puisqu'in vitro on peut fabriquer des vésicules avec des ag seulement, les liaisons hydrogènes de la première composante peuvent se faire et se défaire rapidement localement, tout en maintenant la cohésion globale horizontale. Ce qui compense la grande inertie des queues hydrophobes. Dans les vésicules sans bras mobile,avec des ag seulement, les têtes carboxyliques disposées en tête-bêche peuvent former des liaisons hydrogènes, mais ne forment pas une cohésion globale horizontale.
Aussi, s'il arrive que la liaison ester du bras mobile vienne à être hydrolysée, ou que toute la tête hydrophile vienne à manquer, la place est libre pour toute autre molécule portant une amine. C'est ce que j'appelle la séquestration.
La chiralité des acides aminés séquestrés:
[modifier | modifier le wikicode]Dans le cas des aa séquestrés, la première composante de la cohésion horizontale n'est rétablie entièrement que si le NH2 (ou même un OH) est accompagné d'un P ou d'un carbone carboxylique, comme c’est le cas des aa, pour rattacher l'amine du PLD de l'arrière et qui se trouve à son niveau. Ces aa vont être bien sûr de type lévogyre (L) comme la sérine, puisqu'on se trouve toujours dans la même configuration où il n'y a pas de brèche dans le liposome (trou sans ag) et que les chaînes de PLD se côtoient (voir Dessin 1-RG ci-dessus).
Les autres caractéristiques physico-chimiques des acides aminés séquestrés:
[modifier | modifier le wikicode]L'espace libéré par la tête hydrophile a une surface bien définie. Ça serait là l'origine géométrique de ces aa prébiotiques. Mais leur origine est aussi mécanique puisqu’ils agissent mécaniquement comme la sérine liée au P. Ils ont donc une origine mécanique et géométrique.
- Horizontalité: Ces aa sont disposés horizontalement à l'inverse de ceux qui traversent la membrane dont la disposition pourrait être aléatoire.
- La longueur maximale de ces aa est approximativement équivalente à 7 atomes (voir Dessin 1 ci-dessus), sans tenir compte du carbone carboxylique. Et l'aa ne peut dépasser cette longueur, sinon il empêcherait le glissement des chaînes de PLD voisines et vice-versa.
- La largeur maximum de ces aa, ou épaisseur, est limitée à 4 carbones (voir Dessin 1 ci-dessus). Ceci conduit à de petites chaînes latérales comme avec val, thr, leu, ile, arg, ou à l'épaisseur d'un seul cycle aromatique comme phe, trp, tyr, his.
- En hauteur, c'est-à-dire en dehors de la membrane, il n'y a pas de limite.
Les fonctions chimiques portées par le radical des acides aminés séquestrés:
[modifier | modifier le wikicode]Il n’est pas question ici de retrouver les chaînes des réactions chimiques du réseau métabolique prébiotique. Mais si la séquestration par la cohésion mécanique favorise certains aa et en exclut d'autres, cela nous permettrait d'entrevoir l'initialisation du métabolisme prébiotique à partir de certains groupements d'aa.
- Les acides carboxyliques et les amides:
Il est remarquable de constater qu’il y ait 2 fonctions carboxyliques et 2 fonctions amides courtes à 1 et 2 atomes de plus que la sérine, en longueur, chez les aa biotiques! Pourquoi ces longueurs et ce double emploi unique parmi les 20 aa biotiques? La réponse peut-elle provenir de la cohésion mécanique? On peut effectivement émettre l'hypothèse qu’à ces longueurs ces 2 fonctions se trouvent au niveau du P de la tête perdue et qu’elles puissent attirer l'amine du PLD de l'arrière par liaison hydrogène. Ce qui rétablirait presque entièrement la première composante de la cohésion horizontale comme le ferait un P, alors que la fonction carboxylique de la tête des autres aa, décalée par rapport à l'amine du PLD arrière, ajouterait un déséquilibre à ce rétablissement.
On peut aussi supposer, dans le cas où la tête ne garde que le glycérol du bras fixe, que l'hydroxyle libre de celui-ci puisse établir une liaison hydrogène avec ces fonctions et une liaison ester même avec la fonction carboxylique. Ce qui rétablirait et la première composante horizontale et la composante verticale de la cohésion mécanique.
Par contre une fonction carboxylique ou amide d'un aa plus long arriverait au-dessus des ag, serait repoussée par répulsion électronique et déstabiliserait la fixation de l'aa.
- Les radicaux portant un hydroxyle ou une amine et arrivant au niveau des ag, peuvent établir des liaisons hydrogène avec ces derniers. Ce qui rétablirait partiellement la composante verticale de la cohésion. C'est le cas de arg et tyr (4 atomes de plus que la sérine en longueur), de lys et his (plus 3 atomes).
- Les radicaux hydrophobes, peu réactifs devraient remplir toute la largeur de l'espace libéré. C'est une consolidation par encombrement stérique. C'est le cas de 9 aa biotiques qui sont soit branchés (leu, ile, val, thr), soit aromatiques(phe, trp, pro), soit contenant un atome de soufre(met, cys) qui a autant d'influence stérique par son volume et son noyau que l'atome de phosphore.
- Il n'y a que 2 aa biotiques qui n'apportent que leur tête pour la cohésion mécanique: ala, gly.
- Les problèmes que soulèvent certains aa biotiques tels que met, cys, pro, trp:
Tout radical portant un atome à forte action sur son voisinage devrait être exclu, comme les métaux de transitions, les halogènes, les cations mais aussi le soufre. Dans ce cas la cys peut avoir une origine non mécanique et provenir de la thiolyse de la liaison ester sérine-P, par exemple. Reste l'origine de la met. La pro peut-elle établir une liaison hydrogène sous sa forme cyclique? Ou bien la pro prébiotique était-elle linéaire? Le trp devrait poser un problème de hauteur puisque aucun aa biotique n'a de la hauteur! Est-ce que la citruline et l'ornitine feraient parties des aa prébiotiques séquestrés?
De l'homochiralité prébiotique à l'homochiralité biotique
[modifier | modifier le wikicode] Nous venons de voir les principes théoriques de la mise en place de l'homochiralité, en supposant que les molécules nécessaires sont, soit préexistantes en quantité dans le milieu réactionnel (prébiotique) comme les ag, soit qu’elles puissent être récupérées facilement par déplacement des équilibres thermodynamiques.
Nous allons étudier dans ce chapitre le rapprochement de ces 2 processus en tenant compte des conditions plus contraignantes de l'hypothèse de la poche de pétrole prébiotique et en prenant des circuits métaboliques biotiques les moins complexes possibles. Et cela pour essayer d'imaginer l'évolution moléculaire qui pourrait s'opérer du prébiotique au biotique pour cette mise en place de l'homochiralité.
Les processus prébiotiques pour la mise en place de l'homochiralité
[modifier | modifier le wikicode] Ce sont d’abord les processus fondamentaux de la thermodynamique statistique en milieu liquide (huile ou eau). Les molécules diffusent librement dans le milieu, et interagissent entre elles avec des énergies augmentant avec la température. Dans l'environnement organisé d'une cellule, les petites molécules ne sont pas libres et sont contrôlées et transportées même par des macromolécules. Le cytoplasme est considéré comme un "gel colloïdal".
C'est cette différence fondamentale qui nous interdit de plaquer les réactions enzymatiques du métabolisme biotiques aux réactions chimiques d'un mélange complexe, liquide, sans macromolécules. Le point de départ de l'évolution moléculaire, dans notre hypothèse de poche de pétrole prébiotique, étant l'auto-assemblage des ag synthétisés abiotiquement, notre objectif que nous avons introduit ci-dessus, c’est de mettre en parallèle ces 2 mondes, de les rapprocher en prenant les réactions du métabolisme biotique proches des réactions de la chimie organique, et de considérer les conditions géochimiques de la poche de pétrole les plus proches des caractéristiques physico-chimiques du vivant.
Les conditions de la poche de pétrole prébiotique:
[modifier | modifier le wikicode] Les conditions physico-chimiques les plus proches des caractéristiques du vivant sont celles, par exemple, des bactéries vivants dans les poches de pétrole "fossile", à 55 °C et sous 400-800 bars[3]. Mais on peut s'en éloigner encore un peu pour dépasser légèrement la température maximale de la multiplication des extrêmophiles de 113 °C, à 150 °C; la pression a moins d'influence sur les bactéries et on peut la doubler pour atteindre 1,5 kbar comme dans le gisement de pétrole de Tupi au large du Brésil Tupi.
Je pense que dans ces conditions, les réactions qui s'y déroulent peuvent être en équilibre thermodynamique et donc produire en continu, certaines molécules, si celles-ci sont piégées par ailleurs. C'est le cas de l'hydroformylation qui produit le glycéraldéhyde et la dihydroxyacétone (100-140 °C, 120 bars, à partir de CH2O H2 CO; Györgydeák[4] et al. 1998), de l’hydrogénation du glycéraldéhyde en glycérol (50 °C, 60 bars, en présence de H2) et la synthèse d'aa et d'éthanolamine (A.D.Aubrey et al. (2009), Fig 5[5]) et de la choline (synthèse analogue à la triméthylamine).
Dans l'hypothèse d'une formation du pétrole abiotique par diagénèse (voir pétrole prébiotique), où pression et température s'élèvent progressivement, CH2O H2 CO NH3, issus des clathrates de départ (J.L Charlou[6], Ifremer: Serpentinisation et synthèse inorganique d'hydrogène, méthane et hydrocarbures le long de la dorsale médio Atlantique), pourraient exister et former glycérol, glycéraldéhyde, dihydroxyacétone, éthanolamine, choline ainsi que quelques aa en petites quantités.
Dans l'hypothèse d'une formation brutale du pétrole abiotique (voir pétrole prébiotique) par métamorphisme, lors de la subduction ou proche de ces zones (cônes d'accrétion), les conditions de pression et de température sont au niveau supérieur des limites pour la synthèse du pétrole par le processus FTT. Ces conditions rendraient possible la synthèse du glycérol et de l'éthanolamine directement comme dans les procédés industriels (500 °C et hautes pressions) à partir des molécules d'éthylène et de propène issues du processus FTT. Ces hautes températures ne permettraient pas la formation des têtes hydrophiles. Il faudrait attendre la migration des poches de pétroles vers des zones à températures plus modérées. À ces températures la synthèse de ces 2 molécules, éthylène et propène, ne se faisant plus, il faudrait supposer que de grandes quantités de glycérol et d'éthanolamine aient été synthétisées auparavant, en même temps que les ag et dans les mêmes concentrations, pour subvenir à la formation des têtes hydrophiles. Aucun résultat, dans le domaine du pétrole "fossile" ou synthétique n'a, jusqu'à présent été rapporté dans ce sens, pour le glycérol (pétrole fossile et synthétique, absence de N2) ou pour l'éthanolamine (pétrole fossile, présence de N2).
La mise en place de l'homochiralité prébiotique:
[modifier | modifier le wikicode] Plaçons-nous donc dans le cas d'une poche de pétrole prébiotique formée lentement par diagénèse, où un réseau de réactions chimiques existe en équilibre thermodynamique et qui peut fournir en continu P, glycérol, GA, DHA, éthanolamine, choline, sérine et d'autres aa. Les concentrations de ces molécules n'ont pas d'importance, parce que seul compte la possibilité de piégeage qui déplace les équilibres. Et ce piégeage se fait par les ag et les PLD intégrés dans des macro-structures cohérentes, difficiles à hydrolyser, comme les vésicules dans la phase huile ou les liposomes dans la phase eau.
Cependant pour la fixation des têtes hydrophiles sur les têtes des ag, toutes les réactions sont des estérifications. Or dans l'eau l'hydrolyse tend à les défaire. Et la phase huile, qui permettrait de les conserver, est réduite à l'épaisseur de la bicouche, si cette fixation devait se faire dans des bicouches d'ag de la phase eau (équivalentes des liposomes sans tête hydrophile).
- Les vésicules de la phase huile.
Dans la phase huile les vésicules concentrent toutes les molécules hydrophiles avec en principe peu d'eau. Nous avons vu dans l’article de la chimio-osmose prébiotique, que le liposome dans la phase eau représente le fondement même du vivant, puisqu’il délimite un intérieur spécifique, d'un extérieur changeant et qu’il permet la croissance de l'organisation à l'intérieur par une communication (électronique et ionique) à travers la membrane. Les vésicules de la phase huile, elles, ne peuvent pas communiquer avec l'extérieur, l'huile constituant un mur infiniment épais, ne permettant que l'arrivée des molécules hydrophiles. Les vésicules sont vites isolées les unes des autres.
Par contre les vésicules de la phase huile possèdent une vraie interface eau/huile. Dans une telle interface, les molécules de chimie organique produites pendant le processus de la formation de la poche de pétrole prébiotique, peuvent occuper les 2 phases et créeraient même une phase intermédiaire pour les molécules qui sont plus ou moins solubles dans les 2 premières phases. Cette zone intermédiaire peut être très mince, mais elle permet, avec les 2 autres phases, toutes les réactions possibles suivant cette graduation de la solvatation. Et notamment elle permet l'estérification des têtes des ag par les têtes hydrophiles, l'hydrolyse des liaisons ésters devenant de plus en plus difficile avec l'étendue de la surface phospholipidique.
- Synthèse et fixation par estérification de la tête hydrophile.
D'après ce que nous savons, l'estérification est une réaction réversible; le sens du déplacement de l'équilibre dépend uniquement du solvant; pour un solvant donné l'état d'équilibre ne dépend ni de la température ni de la pression; les réactions sont très lentes en l’absence de catalyseur; le catalyseur est le proton H+ issu de HCl, H2SO4, H3PO4; les têtes carboxyliques sont aussi catalyseurs (auto-catalyse): voir les esters dans wikipédia.
Aussi nous allons supposer que les estérifications qui nous concernent sont possibles dans l'environnement de la poche de pétrole prébiotique et qu’elles se réalisent lentement, et même très lentement étant donné que le temps géologique n’est pas déterminé. Cependant par comparaison avec le métabolisme biotique, nous allons voir dans ce qui suit, que le rôle de celui-ci va agir surtout sur le nombre d'intermédiaires du réseaux des réactions chimiques.
Par exemple le déplacement d'équilibre du mélange racémique du glycérol-P vers le glycérol-3P fixé par les ag, va nécessiter l'enlèvement du glycérol-1P ou du 2P de la surface des ag, leur hydrolyse, puis l'estérification du glycérol libéré en glycérol-3P, puis diffusion de ce dernier vers la surface d'ag. La fixation de ce dernier déplacera alors effectivement l'équilibre du GA en glycérol (réaction d'hydrogénation).
Devant ce grand nombre d'intermédiaires et le potentiel catalytique de la surface des ag et/ou des têtes hydrophiles, il est tout à fait légitime de considérer l'hydrogénation du glycéraldéhyde-P racémique (ou de la dihydroxyacétone) en glycérol-3P après fixation du 1er sur l'ag. Car la tautomérie céto-énolique de la fonction aldéhyde leur permet d'adopter la conformation imposée par la surface, comme nous l'avons vu dans l'approche théorique de la chiralité du liposome (Dessin 1-RG, et Dessin 2). Une fois le glycéraldéhyde-P fixé et la conformation glycéraldéhyde-3P réalisée, alors l'hydrogénation rendue possible par la présence de H2, comme nous l'avons suggérer ci-dessus (Les conditions de la poche de pétrole prébiotique), et par l'influence catalytique de la surface, peut se faire facilement.
Chez le vivant l'inertie engendrée par les déplacements de ces nombreux équilibres est éliminée grâce au transport des petites molécules par les macromolécules, et seule la conformation adéquate est synthétisée et protégée de la racémisation par ces macromolécules. Par ailleurs on conçoit facilement que la mobilité de certaines enzymes dans la phase eau (l'intérieur du liposome ) sera une des 1ères étapes de l'évolution moléculaire, car elles pourront agir successivement à plusieurs endroits de la surface, en emmenant les petites molécules nécessaires avec elles. Sinon, sans cette mobilité, les petites molécules se fixeraient au hasard, puis suivant l'environnement surfacique, la catalyse pourrait être plus ou moins efficace, et les déplacements d'équilibre, en cas d'erreur, rallongeraient énormément la réalisation.
- Formation des liposomes.
Nous avons vu précédemment que les vésicules bicouches d'ag dans l'eau ne conviennent pas pour la formation des molécules de phospholipides. De même la formation des liposomes hétérogènes, formés d'assemblage au hasard de phospholipides et d'ag, comme décrit dans la littérature ( Segré 2000[7], par exemple ), devraient aboutir difficilement, pour les mêmes raisons, à un liposome homogène, ou structuré (têtes sérine, choline et éthanolamine) avec très peu d'ag nus.
Le scénario de la formation des liposomes que je décris ici, suppose que les conditions de température et de pression sont stables sur une longue période, que la poche de pétrole n’est pas soumise, pendant cette période, à des turbulences, permettant ainsi la formation de phospholipides dans les vésicules de la phase huile. L'interface eau/huile des 2 phases globales sera, dans cette hypothèse, constituée, avec la durée, de phospholipides provenant de vésicules ayant migré vers la phase globale eau et y ayant versé leur eau.
Pour qu'une vésicule PLD puisse donner une bicouche PLD, il faut que son feuillet lipidique ne se rompt pas et qu'elle ne rompt pas le feuillet PLD constituant l'interface de la phase globale. Ces vésicules vont s'accumuler en bas de la phase huile, et quand la pression de celles qui sont au-dessus devient assez grande, celles qui sont en contact avec le feuillet global, vont soit éclater comme nous l'avons décrit ci-dessus, soit se détacher pour entrer dans la phase eau globale en s'entourant d'un morceau du feuillet global d'une longueur équivalente à la longueur de son feuillet interne.
Ce scénario de la formation des liposomes donne 2 feuillets homogènes mais structurés différemment avec son feuillet interne et le contenu en eau spécifiques pour chaque liposome.
Hypothèses sur l'évolution moléculaire du processus prébiotique au métabolisme biotique
[modifier | modifier le wikicode]31.01.12
Introduction
[modifier | modifier le wikicode]- Il faut bien préciser encore que nous partons d'un milieu réactionnel aux nombreuses sortes de petites molécules, constituant un liquide en équilibre thermodynamique et entouré d'une surface ionisée; et non d'un gel colloïdal concentré en macromolécules arborant de vastes champs électro-dynamiques liés à leur grande surface portant de nombreuses charges positives et négatives à la fois.
Le point de départ du processus prébiotique de la poche de pétrole abiotique est un mélange liquide en contact avec la paroi ionisée négativement des têtes carboxyliques des ag, neutralisée par un mélange de cations. Cet ensemble doit évoluer vers l'état d'une cellule composée d'un gel colloïdal entouré d'une membrane ionisée négativement et neutralisée par quasiment que des cations K+. Il est intéressant de noter ici que les découvertes, dans les recherches sur les origines de la vie, faites sur le piégeage des molécules organiques (dont les P) par les surfaces minérales, mettent en jeu des surfaces ionisées positivement et neutralisées par des anions Cl-, la plupart des cas.
Dans ce qui suit j’ai privilégié le processus de séquestration des aa et d'autres molécules en plus de la fixation des têtes hydrophiles. Mais il n'y a pas que ce processus qui puisse sélectionner les aa dans l'évolution moléculaire. La séquestration apparaît comme un tri grossier portant sur le rapprochement des 2 fonctions acide et amine de la tête de l'aa, puis sur leur taille limite. Lorsque j’avais émis l'hypothèse de la séquestration des aa, j’avais prolongé ma réflexion pour imaginer les potentialités des aa et certains ont soulevé des problèmes intéressant vis-à-vis de cette hypothèse.
Il est hors de question de ne soumettre la sélection des aa qu'au seul processus de la séquestration tant les caractéristiques pour la sélection de 20 aa sont nombreuses. On peut se poser ainsi un grand nombre de questions pour savoir pourquoi ce nombre quasi immuable et pourquoi ceux-là spécialement.
- J’ai recensé 5 processus qui peuvent déterminer la forme des aa et la fonction chimique de leurs radicaux:
- Diffusion à travers la membrane (voir chimio-osmose prébiotique)
- Séquestration par la cohésion mécanique du liposome
- Acylation des aa aux phosphates des membranes pour l'encrage des protéines à ces membranes.
- Les contraintes des voies de synthèse métaboliques,synthèses qui peuvent se dérouler aussi dans l'eau loin de la paroi.
- Les fonctions chimiques à effectuer, notamment :
- - Emboitement dans la partie hydrophobe de la bicouche lipidique (aa hydrophobes) et en général participation à la structure 3 D des protéines en intéraction avec d'autres protéines et les membranes par liaisons hydrogènes et attraction électrique entre les dipôles électriques (aa polaires et ionisables);
- - Action à distance par les champs électromagnétiques (aa aromatiques) et
- - Participations aux réactions chimiques proprement dites, c'est-à-dire création et suppression des liaisons covalentes (aa réactifs).
- - Emboitement dans la partie hydrophobe de la bicouche lipidique (aa hydrophobes) et en général participation à la structure 3 D des protéines en intéraction avec d'autres protéines et les membranes par liaisons hydrogènes et attraction électrique entre les dipôles électriques (aa polaires et ionisables);
Dans ce qui suit nous allons:
- Illustrer la stratégie mise en œuvre par le vivant pour élaborer les têtes hydrophiles: Au-delà de la chiralité il y a la conformation spatiale en générale, ce qui explique l'intervention des nucléotides CTP et dCTP pour la mise en place du bras mobile.
- Étudier le cas de la chiralité et de l'homochiralité des sucres dont les origines seraient dans la cohésion mécanique des liposomes: l'hydrogénation du L-glycéraldéhyde-3P en Glycérol-3P sur le bras fixe discrimine le L-glycéraldéhyde en faveur du D-glycéraldéhyde.
- Et mettre en œuvre la cohésion mécanique du liposome pour l'initialisation du métabolisme prébiotique: Ici nous étudierons l'initialisation et les étapes de l'évolution du métabolisme prébiotique en considérant en plus la séquestration des aa, de la dCTP et de 3 coenzymes. Ces coenzymes interviendront dans l’ordre suivant: aa prébiotiques, B6, dCTP, B1, CTP et THF.
Au-delà de la chiralité il y a la conformation spatiale en générale
[modifier | modifier le wikicode]
- Pour la notation des enzymes du type (EC 4.3.1.7; 260-1-0), voir figure 1 et 2 ci-dessous.
- Dans les chapitres qui suivent j’ai ajouté les abréviations suivantes, parce que répétitives:
- GA, DGA, LGA pour glycéraldéhyde racémique, D et L
- DHA pour dihydroxyacétone;
- PE, PC, PS pour phosphatidyléthanolamine, phosphatidylcholine, phosphatidylsérine.
- PtdGro, Ptd2Gro pour phosphatidylglycérol et diphosphatidylglycérol (ou cardiolipine).
- Pour la notation des enzymes du type (EC 4.3.1.7; 260-1-0), voir figure 1 et 2 ci-dessous.
- Pour rechercher les enzymes dans KEGG cliquez ici: [1]
- Pour rechercher les enzymes dans KEGG cliquez ici: [1]
- La chiralité du glycérol-P est établie pendant:
- - l'hydrogénation de la glycérone-P, molécule achirale favorisant la tautomérie;
- - la phosphorylation du glycérol, molécule achirale par symétrie, permettant de fixer le phosphate après la mise en place du glycérol.
- La fixation de la sérine permet de positionner son NH2, comme on l'a vu dans le dessin 1-RG. Elle constitue le seul bras mobile portant une fonction très réactive, la fonction carboxylique.
- La fixation du glycérol en tant que bras mobile, illustre admirablement la nécessité de bypasser la sélection de la conformation adéquate parmi une multitude d'autres conformations. Cette stratégie est utilisée aussi pour la création du bras éthanolamine après fixation de la sérine, comme nous allons le voir dans ce qui suit.
- La création de la PE ne passe pas par la fixation de l'éthanolamine libre. En plus du positionnement de NH2 par la sérine, la PE illustre bien l'importance de la conformation spatiale en général par le fait que l'éthanolamine n'a pas de carbone assymétrique, mais aussi par la nécessité d'éliminer toute entrave à la cohésion mécanique globale du liposome. Parce que l'éthanolamine libre, une molécule très petite, peut échapper au contrôle des macromolécules et mal se positionner ou même établir des liaisons hydrogène avec les acides gras détruisant la cohésion globale.
- Le liposome, en effet, n’est pas rigide. Toutes les molécules PLD de la bicouche sont en mouvement perpétuel tout en assurant la cohésion de l'ensemble. Chez les procaryotes, en effet, l'éthanolamine n'intervient que dans une seule réaction (EC 4.3.1.7; 260-1-0), celle qui la détruit en acétaldéhyde et en NH3. Chez les eucaryotes elle est balisée par une plus grosse molécule qu'est le CDP (EC 2.7.1.82; 2.7.7.14; ) . Ce qui l'empêche de devenir un bras mobile mal emboité. Chez les eucaryotes, autres que les champignons (EC 2.7.8.8; 860-70), la PS est synthétisée à partir de la PE et la sérine (EC 2.7.8.29;0-0-40 ).
- Il est remarquable de noter que la choline libre, avec sa grosse tête de triméthylamine (EC 2.7.8.24; 100-0-0), est utilisée directement sans balise pour synthétiser la PC chez de nombreux procaryotes mais chez aucun eucaryote, comme la sérine le fait avec la PS chez tous les procaryotes et les champignons mais pas chez les eucaryotes supérieurs. Par ailleurs la choline se différencie encore de l'éthanolamine par le fait qu'elle n’est pas détruite immédiatement, mais qu'elle est dégradée en plusieurs étapes en glycine.
- Mais chez tous les êtres vivants la synthèse du bras mobile passe par une estérification intermédiaire inattendue par la CTP. C'est une différence essentielle avec la théorie du métabolisme prébiotique basée sur l'estérification directe des petites molécules. Et cette étape intermédiaire regroupe tous les fondamentaux du métabolisme biotique à savoir un nucléotide (CTP), un désoxynuléotide (dCTP) avec un rapport CTP/dCTP égale à 0.88 (dans Biochemistry of lipids, lipoproteins and membranes 2008, page 74[8]), un phospholipide et bien sûr une enzyme. Mais ne fait pas intervenir de coenzymes, ni CoA ni B6. Nous avons là réunis en monomères, l'ADN, l'ARN, une protéine et la membrane. Nous allons voir dans ce qui suit que cette étape d'estérification a une seule raison d'être, c’est de mettre en place correctement le bras mobile pour qu’il puisse remplir sa fonction principale, la cohésion mécanique du liposome.
-
 Figure 1. Ce diagramme représente la compilation des organismes possédant un gène donné de la voie métabolique des glycérophospholipides: D'après une impression d'écran de la base de données des voies métaboliques de KEGG.
Figure 1. Ce diagramme représente la compilation des organismes possédant un gène donné de la voie métabolique des glycérophospholipides: D'après une impression d'écran de la base de données des voies métaboliques de KEGG.
Les rectangles colorés correspondent à des enzymes membranaires. La compilation est un comptage personnel et approximatif des organismes listés dans la base et consiste en 2 nombres, le 1er celui des procaryotes et le second aux eucaryotes.
Quand l'effectif est faible un 2e comptage des listes des bases Brenda ou RefSeq (à partir des liens indiqués par KEGG) est accolé à celui issu de la liste de KEGG. Exemple: 0.5.60 correspond à 0 procaryotes dans la liste de KEGG, 5 procaryotes dans Brenda et RefSeq, et 60 eucaryotes dans KEGG. Les dessins des flèches et les noms des molécules sont ceux de KEGG. -
 Figure 2. Analogue à la figure1, d’après les voies métaboliques de KEGG. Ici c’est un montage d’après plusieurs voies métaboliques pour représenter la circulation entre la Sérine, la Glycérone, les acides gras et les phospholipides : Fatty acid Biosynthesis, Fatty acid Metabolism, Glycerolipid Metabolism, Glycerophospholipid Metabolism et Glycolysis.
Figure 2. Analogue à la figure1, d’après les voies métaboliques de KEGG. Ici c’est un montage d’après plusieurs voies métaboliques pour représenter la circulation entre la Sérine, la Glycérone, les acides gras et les phospholipides : Fatty acid Biosynthesis, Fatty acid Metabolism, Glycerolipid Metabolism, Glycerophospholipid Metabolism et Glycolysis.


La cohésion mécanique du liposome est à l'origine de la chiralité et de l'homochiralité des sucres
[modifier | modifier le wikicode]Dans le métabolisme biotique tous les sucres ont la configuration D pour le 1er carbone asymétrique qui suit la fonction aldéhyde ou cétonique. C'est le cas notamment du D-Glycéraldéhyde à la base de la synthèse de tous les sucres biotiques. Or dans la théorie de la cohésion mécanique du liposome nous n'avons démontré que l'homochiralité du glycérol-3P, de la sérine et éventuellement des autres acides aminés si leur séquestration par les liposomes est effective. En plus le DGA ne fait pas partie de la bicouche lipidique et n'intervient qu'en second lieu à la synthèse du glycérol-3P.
Comment alors s'est établi l'homochiralité du DGA? Peut-on faire intervenir la cohésion mécanique du liposome? Si c’était le cas, il faudrait que le glycéraldéhyde soit séquestré comme les aa, c'est-à-dire prendre la place du bras mobile de la tête hydrophile et qu’il soit de chiralité L. C'est donc le LGA qui aurait pu être séquestré et avantagé par rapport au DGA. Comment alors le LGA a-t-il pu disparaître complètement dans l'évolution moléculaire pour laisser place au seul DGA? Une fois fixé le LGA peut-il être hydrogéné en glycérol? Mais il faut alors supposer une hydrogénation différente de celle du bras fixe.
- La disparition du LGA du métabolisme biotique est due à sa fixation sur le bras fixe en compétition avec la DHA.
- Nous avons vu au chapitre chiralité du glycérol-P que pour les bactéries le glycérol-P qui fixe les acides gras est un L-glycérol-3P. Il a la même conformation que le LGA-3P avec un aldéhyde à la place de l'alcool terminal. Donc le LGA peut prendre la place de la DHA. Ensuite il y a hydrogénation pour obtenir du glycérol-3P, avec la tautomérie du LGA (comme on l'a supposée pour la glycérone) pour faciliter et permettre, dans un premier temps, la configuration spatiale adéquate.
- Du coup l'élimination du LGA entraîne, sinon sa disparition totale (suivant le degré de réversibilité de l'isomérisation prébiotique L <--> D du GA), du moins son désavantage sélectif par rapport au DGA qui restera libre et non fixé aux PLD. L'homochiralité des sucres peut alors commencer. Nous voyons ainsi que l'homochiralité des aa provient de leur séquestration ou de leur fixation (sérine) dans les bras libres des PLD et se fait encore dans le métabolisme biotique, alors que l'homochiralité des sucres a dû se faire une fois seulement pendant le métabolisme prébiotique, le LGA disparaissant après complètement.
- Chiralité du PtdGro
- Le glycérol-P du bras mobile de PtdGro est le L-glycérol-3P. En effet il a même configuration que la L-sérine avec l'alcool terminal (CH2OH) à l'extérieur de la membrane. C'est le même que celui du bras fixe puisque celui-ci a l'alcool terminal disposé vers l'intérieur de la membrane (paragraphe chiralité du glycérol-P). PtdGro est décliné, dans Kegg, en phosphatidylglycérol sans préciser la chiralité du glycérol. À mon avis il faut le nommer phosphatidyl-L-glycérol (sans ajouter 3P qui porterait à confusion). D'ailleurs la réaction est écrite clairement dans Kegg[9]:
- CDP-diacylglycerol + sn-glycerol 3-phosphate = CMP + 3(3-sn-phosphatidyl)-sn-glycerol 1-phosphate (EC 2.7.8.5; 1200-110)
- le P de sn-glycérol 1-P se réfère au P terminal, alors que le substrat est bien sn-glycérol 3-P.
- CDP-diacylglycerol + sn-glycerol 3-phosphate = CMP + 3(3-sn-phosphatidyl)-sn-glycerol 1-phosphate (EC 2.7.8.5; 1200-110)
- Le PtdGro existe aussi dans les têtes hydrophiles des Archées[10]. On a vu que chez les bactéries que le L-glycérol-3P pouvait venir de l'hydrogénation de DHA ou du LGA-3P. Si pour les archées le D-glycérol-3P devait provenir, au début de l'évolution prébiotique, aussi du DGA-3P on aurait eu une homochiralité L de tous les sucres. Ce qui n’est pas le cas. Donc les archées, ayant tous leurs sucres de chiralité D, sont apparus après les bactéries.
- Il est à noter cependant que la synthèse du D-glycérol-3P et son utilisation restent toujours possibles même dans le métabolisme biotique des bactéries. En effet le Ptd2Gro (EC 278.-, Cls;700-100), ubiquitaire, a un glycérol flanqué de 2 acides phosphatidiques avec 4 liaison esters hydrolysables. La libération du glycérol-P peut se faire de 2 manières possibles donant chacune un glycérol-3P ou un glycérol-1P (dans Biochemistry of lipids, lipoproteins and membranes 2008, page 75)[8]).
- On aurait tendance à dire que les premiers bras mobiles qui seraient apparus, aient été des L-glycérol-3P puisque ceux-ci ont été déjà synthétisés à la première étape de la constitution de la tête hydrophile. Mais il faut remarquer que la synthèse du PtdGro demande une étape de plus que celle du PS, à partir d'un glycérol qu’il faut détacher du bras fixe; que le glycérol risque, s'il était l'unique bras mobile, d’établir dans certaines conditions de température et de pression des liaisons hydrogènes généralisées, puisqu’il n’est pas ionisable, ce qui cristallisera le liposome.On voit alors l'importance de la sérine qui peut être synthétisée abiotiquement[5], même en petite quantité, ou bien qu'elle puisse être synthétisée en une seule réaction enzymatique (en présence de B6), dans le métabolisme biotique (EC 431.17; 800-20, et 431.19; 930-100). La sérine est intéressante parce que c’est un acide aminé réactif par sa tête zwitterionique et qu'elle peut en une seule étape donner PE dont l'amine est plus réactive, encore, que le glycérol.
La CMP positionne le tétraèdre du phosphate pour estérifier le bras libre dans la bonne configuration nécessaire à la cohésion mécanique du liposome.
[modifier | modifier le wikicode]- Nous avons vu au paragraphe sur la conformation spatiale en générale, la différence essentielle et inattendue entre estérifications prébiotiques hypothétiques et les estérifications biotiques. En considérant la configuration spatiale en général au lieu de la chiralité seule, il apparaît clairement qu'en prébiotique, le nombre de configurations explose lorsqu'on tient compte de la rotation du phosphate libre avant son estérification par la sérine, le glycérol, l'éthanolamine ou la choline.
- Dans la fixation du glycérol du bras libre, en métabolisme biotique, le phosphate (du glycérol-3P) a servi à le baliser et peut-être à forcer sa fixation dans la position adéquate de la cohésion mécanique. Mais le P du PtdGro-P alourdit le bras mobile (EC 2.7.8.5; 1200-110). Le phosphate est expulsé rapidement dans la réaction EC 3.1.3.27 (500-4-0) chez la plupart des procaryotes.
- Il y a 3 enzymes qui positionnent le phosphate, (Ec 2.7.7.41; 1200-140) pour la sérine et le glycérol, (Ec 2.7.7.14; 0-0-20) pour l'éthanolamine et (Ec 2.7.7.15; 25-70) pour la choline. Nous voyons que seule la première est généralisée à tous les êtres vivants, alors que les 2 autres sont plutôt propres aux eucaryotes seulement.
La cohésion mécanique du liposome et l'initialisation du métabolisme prébiotique
[modifier | modifier le wikicode]Remettons-nous dans les conditions de la poche de pétrole prébiotique comme on l'a décrit dans les conditions de la poche de pétrole prébiotique, et reconsidérons l'initialisation du métabolisme prébiotique à la lumière des résultats obtenus jusqu'ici dans une vésicule aqueuse de la phase huile.
Nous avions admis que l'initialisation du métabolisme ne pouvait démarrer qu’à conditions que les molécules nécessaires puissent être piégées par la paroi de la vésicule, déplaçant régulièrement les équilibres thermodynamiques du contenu aqueux. Excéptée l'hydrogénation du DHA ou du GA par H2, hydrogénation dont on avait supposé l'existence, toutes les autres réactions de la synthèse des têtes hydrophiles sont des estérifications très lentes si elles se font sans enzymes. Ce sont dans l'ordre:
- - Phosphorylation du DHA et du GA;
- - Estérification de ces molécules phosphorylées aux ag après hydrogénation in situ, catalysé par le phosphate et la surface carboxylique.
- - Fixation de l'éthanolamine ou de la sérine ou du glycérol. En supposant que ce dernier puisse exister, par exemple par son hydrolyse de la paroi.
- Les résultats de la réflexion sur la théorie de la cohésion mécanique du liposome montre que celle-ci crée un cercle vertueux: l’augmentation du nombre de têtes hydrophiles de même chiralité augmente la cohésion de la vésicule qui favorise à son tour la fixation de molécules de même chiralité, formant de nouvelles têtes hydrophiles.
- Ce cercle vertueux de la cohésion mécanique va aussi créer un autre cercle vertueux basé sur la catalyse: la L-sérine des têtes hydrophiles a son carboxyle libre et très réactif; la cohésion mécanique par ailleurs concentre de plus en plus de L-aa sur une surface de plus en plus en grande de têtes hydrophiles, ce qui favorise le regroupement d'aa en entités catalytiques de plus en plus efficaces.
- Certaines études (Wieczorek, 2009[11]) ont montré par exemple que le dipeptide Ser-His, attaché à la paroi, est très réactif et est à l'origine de nombreux processus qui se déroulent dans le liposome. On le trouve dans les sites actifs de nombreuses enzymes. Dans le cas du métabolisme prébiotique où les réactions sont très lentes, une seule molécule de ce dipeptide peut avoir une forte accélération de l'évolution moléculaire.
- Séquestration de certains coenzymes.
- Le processus de séquestration devient réellement intéressant avec la séquestration de précurseurs de quelques coenzymes (voir leurs dessins ci-dessous) comme la pyridoxamine pour le pyridoxal-P, le noyau pyrimidinique de la thiamine, le noyau pteridinique de l'acide folique et la désoxycytidine. Ce sont de petites molécules de la taille des aa avec une amine et un hydroxyl comme l'éthanolamine. En plus la désoxycytidine se fixe carrément au PLD, sous forme dCMP, comme la sérine dans le métabolisme biotique.
- L'analogie des 3 autres coenzymes avec la dCTP est encore plus frappante puisque leurs intermédiaires sont phosphorylés avant d’être convertis en leur forme active, comme la desoxycytidine: c’est ainsi que le noyau pyrimidinique de la thiamine (EC 271.49, 2747) et le noyau pteridinique du folate (EC 2763) sont phosphorylés avec 2 phosphates dans le métabolisme biotique. Curieusement la pyridoxamine, quant à elle, est convertie en la forme active, le pyridoxal-5P chez Clostridium kainantoi par une transaminase (EC 261.54) à partir du D-Ala ou du D-Glu. Une transaminase de ce type se fait normalement avec un L-aa et a comme coenzyme le pyridoxal-5P.
- Nous allons supposé que dans le métabolisme prébiotique, ces précurseurs sont d’abord séquestrés (ou fixés?) sur la paroi avant d’être convertis en leur forme active. Notamment nous allons voir (tableau 1 étape 5) que la dCTP primordiale pour le métabolisme biotique pourrait être intéressante de l'introduire avant l'apparition de la CTP puisque celle-ci nécessite pour sa synthèse l'ATP (étape 6), alors que la dCTP pourrait apparaitre avant (étape 4).
-
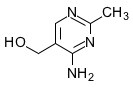 4-Amino-5-hydroxymethyl-2-methylpyrimidine
4-Amino-5-hydroxymethyl-2-methylpyrimidine -
 Thiamine
Thiamine -
 4-Methyl-5-(2-hydroxyethyl)-thiazole
4-Methyl-5-(2-hydroxyethyl)-thiazole -
 desoxyCytidine
desoxyCytidine


-thiazole.png)

-
 Acide folique
Acide folique -
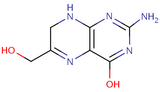 2-Amino-4-hydroxy-6-hydroxymethyl-7,8-dihydropteridine
2-Amino-4-hydroxy-6-hydroxymethyl-7,8-dihydropteridine -
 Acide 4-Aminobenzoïque
Acide 4-Aminobenzoïque -
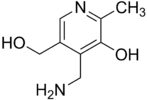 Pyridoxamine
Pyridoxamine -
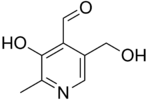 Pyridoxal
Pyridoxal





- Les étapes de l'initialisation du métabolisme prébiotique, Tableau 1:
Nous allons maintenant dérouler le scénario de l'initialisation du métabolisme prébiotique en commençant par la synthèse des têtes hydrophiles générant la cohésion mécanique et la différenciation entre la paroi riche en L-aa séquestrés et l'intérieur liquide riche en D-aa.
Puis interviendrait la séquestration de B6 qui jouerait le rôle de coenzyme pour les groupes de L-aa de la paroi. Je fais intervenir B6 en 1er par analogie, parce que dans le métabolisme biotique, elle active d'innombrable réactions entre les aa plus NH3. Décuplant ainsi le cercle vertueux de la catalyse.
Tableau 1. La cohésion mécanique et les étapes de l'initialisation du métabolisme.
| Fixation | Séquestre | Produits | Cumul | Disparition | Utilisation | aa présents | Commentaires à partir du site KEGG | ||
|---|---|---|---|---|---|---|---|---|---|
| (analogie des groupes de L-aa séquestrés avec les enzymes). | |||||||||
| 0 | Vésicule. Energétique prébiotique : Formose. Synthèse hydrothermale des aa : ADEGS. | ||||||||
| (pour l'analogie avec la synthèse des L-aa voir le site KEGG). | |||||||||
| 1 | DHA LGA L-ser | Têtes hydrophiles PS | DGA D-ser cohésion | LGA | H2 | ADEGS | Hydrogénation catalytique prébiotique, non enzymatique, de DHA et LGA et non du DGA. Accumulation de D-ser et l'homochiralité des sucres est due à la fixation du LGA. | ||
| 2 | L-aa | groupes L-aa | D-aa | L-ser libre initial | ADEGS | ADEGS | Différenciation entre surface (L-aa) et intérieur aqueux (D-aa). | ||
| 3 | B6 | L-ser NQCTWY ADEG PE PtdGro Ptd2Gro |
cohésion 2-oxo-acides |
aa libres initiaux | H2S NH3 indol phenol acetaldéhyde | NQCTWY ADEGS | B6 et le regroupement des L-aa homogènes catalysent mieux les réactions à 1 étape sans ATP, utilisant NH3. Les interconversions entre les aa accumulent les oxo-acides qui avec DHA et DGA ils préparent le métabolisme intermédiare des acides carboxyliques. | ||
| 4 | dR-1P dATP | DGA-3P+acetal Adenine | Réactions sans coenzymes : 4124, 5427 donnent le D-dRibose-1P. Puis avec Adénine : 2421,271.76,2743,2746 donnent dATP. | ||||||
| 5 | CTP | cytosine | cytosine+dR-1P | Cohésion | Cytosine dR-1P dATP | L'équivalent de 2421 n'existe pas pour la cytosine ; les groupes de L-aa pourraient catalyser la synthèse de dCTP sur place. | |||
| 6 | B1 | ATP CTP NAD B6 SAM FAD FMN Biotine FHKPIVM PC |
Cohésion | H2 initial B6 initial | Bases nicotinate DHA+DGA dATP | FHKPIVM NQCTWY ADEGS | B1 est constituée de m1 et m2, dont m1 peut être séquestrée. La synthèse de B1 peut se faire sur place comme pour dCTP. Avec B1, 412.13, 313.11, 2211, 5131, 5316 et à partir de DHA+DGA on obtient R-5P qui avec dATP donne PRPP (2761) et R-1P (5427). A+R-1P+PRPP+PPP → ATP : 2421, 2428, 2743, 2741. C+R-1P+ATP → CTP : 2422, 271.48, 274.14, 2746. N+PRPP+ATP+NH3 → NAD : 242.11, 2771, 6351. D-Ribulose-5P (5131)+DGA-3P+L-gln → Pyridoxal-P (B6) : 4.-.-.-, YaaD, Pyridoxal biosynthesis lyase pdxS. 2.6.-.-, YaaE, Glutamine amidotransferase subunit pdxT. | ||
| 7 | THF | CoA acides gras LR | DHA DGA de formose Phosphate | LR FHKPIVM NQCTWY ADEGS | THF est constitué de m3 et m4 qui peuvent être séquestrées. Pyruvate+B1+NAD+THF+ATP+L-asp+L-cys → CoA : 2216, 11.86, 4219, 212.11, 111.169, 411.11, 6321, 271.33, 6325, 411.36, 2773, 271.24. | ||||
| . | |||||||||
| 8 | Formation des liposomes : passage à l'énergétique membranaire. | ||||||||
| . | |||||||||
| 9 | DHA DGA Bases nicotinate indol phenol | Bases nicotinate indol phenol initiaux | Metabolisme actuel | L-aa actuels | Voir site KEGG | ||||
| 10 | B1 THF | B1 THF initiaux | Metabolisme actuel | L-aa actuels | Voir site KEGG | ||||
- Etape 0.
Ce sont les produits de départ dans la vésicule aqueuse dans la phase huile. Toute molécule est susceptible d’être présente. Cependant pour les concentrations je me réfère aux expériences à haute température (150 °C) et hautes pressions (300 bars).
- - H2 H2S CO2, N2 puis NH3 les gaz des évents hydrothermaux (Charlou[12] 2002, Proskurowski[13] 2008 ).
- - Phosphates et polyphosphates des fonds marins (Arrhenius 1997[14]).
- - alcanes, acides gras, alcools et aldéhydes du processus Fischer-Tropsch, dont l'acétaldéhyde (Rushdi[15] 2001, McCollom 1999[16], 2006[17]).
- - DHA GA glyoxal par hydroformylation ou formose à 120 bars et 140 °C[4].
- - ADEGS aa obtenus dans les expériences hydrothermales avec des molécules azotées[5].
- - Précurseurs des coenzymes et noyau aromatiques en très petites quantités de la poche prébiotique (hypothèse):
- Bases nucléiques, A G C U T, pyridoxamine, les 2 noyaux de la thiamine, les 2 noyaux du folate et le nicotinate pour le NAD.
- Etape 1.
- Synthèse de la tête hydrophyle suivant l'analyse des paragraphes précédent. Fixation sur la paroi de L-ser, DHA et LGA, accumulation de DGA et D-ser dans l'eau. Origine de l'homochiralité des sucres via DGA. Cercle vertueux synthèse_des_têtes/cohésion_mécanique.
- Etape 2.
- - Séquestration des L-aa par les parois et concentration des D-aa dans l'eau. L-ser disparait petit à petit de l'eau avec la diminution de la synthèse hydrothermale des aa avec le temps.
- - Regroupement des L-aa sur la paroi pour constituer des pseudo-enzymes. Elles ne sont peut-être pas très efficaces, mais de multiples combinaisons plus ou moins éphémères sont possibles. Plus la surface des têtes hydrophiles grandit plus les groupes pourraient être solides et nombreux, plus ils seront coopératifs dans la catalyse et leur regroupement même. C'est le cercle vertueux de la catalyse soutendu par la cohésion mécanique.
- Etape 3.
- - L'accumulation de DGA et de D-ser déplacent très lentement les équilibres vers L-ser.
- - La séquestration de B6 accélère l'isomérisation de D-ser en L-ser et la désamination de D-ser en pyruvate (Ec 431.18) puis son amination en L-ser (EC 431.17).
- - Sous l'action de B6, synthèse de nouveaux aa: à partir du pyruvate, du NH3 et de l'indole (Trp) ou du phénol (Tyr); avec la thiolyse par H2S des têtes hydrophiles (Cys); avec la condensation de la Gly et de l'acétaldéhyde en Thr; et l'amination du Glu en Gln et Asp en Asn (voir KEGG pour l'analogie avec les enzymes).
- - Sous l'action de B6, décarboxylation de la sérine de PS pour donner PE (EC 411.65). L'éthanolamine très peu produite dans la synthèse hydrothermale des aa, a pu être fixée à la place de la sérine mais beaucoup plus difficilement comme on l'a vu précédemment. Il y a renforcement de la cohésion mécanique par la PE car elle n'a pas de tête réactive.
- - Sous l'action de B6, désamination de Thr pour donner le 2-oxo-butanoate (EC 4125) nécessaire plus loin pour la synthèse de Val Leu Ile.
- - Transaminations entre les aa (ADEGS plus les nouveaux aa, CNQTWY) pour la production de L-ser, ce qui accumule les 2-oxo-acides.
- - DGA, DHA, 2-oxo-acides, NH3, H2S et les aa ADEGS+CNQTWY constituent le métabolisme intermédiaire prébiotique.
- - Tous ces nouveaux produits grâce à B6 s'obtiennent en une seule réaction.
- Etape 4.
- - Synthèse sans coenzyme du premier desoxy-pentose en 2 réactions (EC 4124, 5427[18]) dont la 2e est auto-catalytique (formation de ribose biphosphate): DGA-3P + acétaldéhyde = D-dRibose-1P.
- - Synthèse de la désoxy-Adénosine sans coenzyme (EC 2421), puis 2 phosphorylations avec P et PPP donnent la dADP (EC 3135, 2743).
- - Le passage à la dATP nécessite l'ATP dans le métabolisme biotique (EC 2746). Je suppose qu'en prébiotique qu’il est tout à fait possible que la dATP se forme très lentement par auto-catalyse (en confondant dATP et ATP dans EC 2746) ou en présence de polyphosphates comme les 2 réactions qui la précèdent. D'ailleurs c’est une estérification et j’avais basé l'évolution moléculaire prébiotique sur les estérifications (voir paragraphe 5.1.2).
- Etape 5.
- - Cette étape peut paraitre théoriquement superflue, mais l'étape 6 qui suit nécessite la thiamine et 7 réactions pour arriver au Ribose-1P. Or nous avons vu à la séquestration de la thiamine, du folate et de la dCTP qu’il fallait une condensation in-situ des 2 parties pour former chaque coenzyme. La dCTP devrait paraitre bien avant la CTP.
- - Dans le métabolisme biotique, il n'existe pas d'équivalent pour la dCTP de la EC 2421 de l'étape 4. Or le passage par la dCTP ou la CTP pour la fixation du bras mobile (EC 277.41), quel que soit le bras, semble primordiale. Aussi la formation in-situ en présence de groupement de L-aa séquestrés apporterait rapidement une grande cohésion mécanique. La dCTP se comporte comme un coenzyme puisque la dCTP, après hydrolyse peut être regénérée par phosphorylation de la dCMP.
- Etape 6.
- - Synthèse in-situ de B1 (voir le dessin des molécules ci-dessous).
- - C'est l'accumulation de DGA dès la première étape du scénario qui va favoriser, par les déplacements des équilibres thermodynamiques, sa condensation avec la DHA, toutes les 2 sous forme phosphorylée: DGA-3P + DHA-P (EC 412.13).
- - ATP: Il faut 6 réactions et la participation de dATP et de B1, seules coenzymes, pour arriver à la molécule centrale PRPP puis une isomérisation pour arriver au Ribose-1P nécessaire à la synthèse de l'ATP à partir de l'Adénine et de PPP.
- - La synthèse de la CTP nécessite l'ATP.
- - Le NAD nécessite le nicotinate et l'ATP.
- - La synthèse de la S-adénosyl-méthionine (SAM), des flavines (FAD et FMN) et de la biotine ne nécessite que les coenzymes prébiotiques déjà créées.
- - Le Pyridoxal-5P, B6, part d'un intermédiaire de la synthèse du D-Ribose-1P, le D-Ribulose-5P: D-Ribulose-5P+DGA-3P+L-Gln (EC 5131).
- - La cohésion va se développer à grande vitesse avec la CTP; remplacement de l'hydrogénation prébiotique (H2) par l'hydrogénation par le NAD et le B6 est synthétisé de novo.
- - 7 nouveaux aa peuvent être synthétisés dont l'histidine qu'on a vu, importante pour les sites actifs des protéines: FHKPIVM.
- - La synthèse de la méthionine permet la synthèse de SAM et de là la production de PC.
- Etape 7.
- - Synthèse in-situ du folate (THF) (voir le dessin des molécules ci-dessous).
- - Synthèse du coenzyme A, CoA et des 2 derniers aa, LR, synthèse des ag.
- Etape 8.
- - La formation du liposome avec le passage de la vésicule dans la phase eau, comme on l'a vu est nécessaire parce qu’à un moment ou un autre, l'énergie prébiotique limitée à la vésicule (formose: DHA et DGA), s'épuisera. L'énergie prébiotique sera remplacée par l'énergétique membranaire qui suit l'évolution moléculaire supposée dans l’article de la chimio-osmose prébiotique.
- - Les molécules de base du métabolisme, notamment le phosphate, arrivent alors par diffusion d’abord, par flip-flop physique pour le phosphate, puis par les porines primitives équivalentes aux ionophores et enfin par les transporteurs protéiques.
- - Les complexes énergétiques protéiques s'installent dans la membrane par migration des aa de l'intérieur ou par diffusion de ceux venant de l'extérieur.
- Etape 9.
- - Avec le métabolisme membranaire, synthèse de DHA et DGA de novo, des acides gras et des coenzymes membranaires comme les cytochromes. Remplacement des noyaux aromatiques: indol, phénol, nicotinate et les bases nucléiques.
- Etape 10.
- - Remplacement de la Thiamine et du folate (THF).
J'arrête ici l'évolution moléculaire du métabolisme. Une autre réflexion devrait s'intéresser à part entière à l'évolution des macromolécules à partir de ce métabolisme qui lui fournirait les monomères.
Références
[modifier | modifier le wikicode]- ↑ http://link.springer.com/article/10.1007/s11084-015-9416-7?sa_campaign=email/event/articleAuthor/onlineFirst
- ↑ http://www.genome.jp/dbget-bin/www_bget?C00093
- ↑ Dorota Wolicka, Andrzej Borkowski, and Dariusz Dobrzynski: Interactions between Microorganisms, Crude Oil and Formation Waters. Geomicrobiology Journal, 27:43–52, 2010.
- ↑ 4,0 et 4,1 Zoltán Györgydeák,István F. Pelyvás. Monosaccharide sugars: chemical synthesis by chain elongation, degradation...(Page 8). Academic Press 1998.
- ↑ 5,0 5,1 et 5,2 A. D. Aubrey & H. J. Cleaves & Jeffrey L. Bada: The Role of Submarine Hydrothermal Systems in the Synthesis of Amino Acids. Orig Life Evol Biosph (2009) 39:91–108 DOI 10.1007/s11084-008-9153-2
- ↑ http://www.ifremer.fr/serpentine/fiches/fiche8.htm
- ↑ Daniel Segré and Doron Lancet: Composing life. EMBO Reports vol.1 no.3, pp 217–222, 2000. http://ool.weizmann.ac.il/Segre_Lancet_EMBOrep_2000.pdf
- ↑ 8,0 et 8,1 Biochemistry of lipids, lipoproteins and membranes 5th edition, 2008: edited by D.E. Vance and J.E. Vance; Elsevier
- ↑ http://www.genome.jp/dbget-bin/www_bget?ec:2.7.8.5
- ↑ Hiromi Daiyasu et al. : A study of archaeal enzymes involved in polar lipid synthesis linking amino acid sequence information, genomic contexts and lipid composition. Archaea 1, 399–410 © 2005 Heron Publishing—Victoria, Canada
- ↑ Gorlero M, Wieczorek R, Adamala K, Giorgi A, Schininà ME, Stano P, Luisi PL. (2009) Ser-His catalyses the formation of peptides and PNAs. FEBS Lett. 583(1):153-6.
- ↑ Charlou J.L., Donval J.P., Fouquet Y., Jean-Baptiste P., Holm N., « Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the Rainbow hydrothermal field », Chemical Geology, vol. 191, 2002, p. 345-359. sciencedirect
- ↑ Giora Proskurowski, Marvin D. Lilley, Jeffery S. Seewald, Gretchen L. Früh-Green, Eric J. Olson,1 John E. Lupton, Sean P. Sylva, Deborah S. Kelley: Abiogenic Hydrocarbon Production at Lost City Hydrothermal Field . Science vol 319, 1 février 2008 sciencemag
- ↑ G. Arrhenius, B. Sales, S. Mojzsis and T. Lee : Entropy and Charge in Molecular Evolution-the Case of Phosphate Journal of Theoretical Biology Volume 187, Issue 4, 21 August 1997, Pages 503-522 sciencedirect
- ↑ Ahmed I. Rushdi, and Bernd R.T. Simoneit: Lipid formation by aqueous fischer-tropsch-type synthesis over a temperature range of 100 to 400 °C. Origins of Life and Evolution of Biospheres (2001) 31: 103–118.
- ↑ T.M.McCollom et al. 1999: Lipid synthesis under hydrothermal conditions by fischer-tropsch-type reactions . Origins of Life and Evolution of the Biosphere 29: 153–166, 1999
- ↑ T.M.McCollom et al. 2006:Carbon isotope composition of organic compounds produced by abiotic synthesis under hydrothermal conditions. Earth and Planetary Science Letters Volume 243, Issues 1-2, 15 March 2006, Pages 74-84
- ↑ http://www.genome.jp/kegg/pathway/map/map00030.html